Erythema Multiforme
Definition:
Erythema multiforme is an immune-mediated illness that usually resolves on its own. It affects the skin and mucous membranes, and is characterized by the presence of “target” lesions. Erythema multiforme major can be distinguished from multiforme minor by the presence of significant mucosal involvement. Episodes can manifest as either solitary, recurrent, or persistent.
Etiology:
1-Approximately 90% of cases are triggered by infection, with HSV type 1 being the predominant cause. Additional infectious triggers include:
- HSV type 2
- Cytomegalovirus
- Epstein-Barr virus
- Influenza virus
- Vulvovaginal candidiasis
- SARS-CoV-2
- Orf.
2-Medications that can potentially cause erythema multiforme include:
- Antibiotics (including erythromycin, nitrofurantoin, penicillins, sulfonamides, and tetracyclines)
- Anti-epileptics
- Non-steroidal anti-inflammatory drugs
- Vaccinations (most common cause in infants).
3-Additional disorders that are commonly linked to erythema multiforme, particularly in cases of chronic disease, include:
- Inflammatory bowel disease
- Hepatitis C
- Leukaemia
- Lymphoma
- Solid organ cancer malignancy
Clinical features:
Cutaneous features:
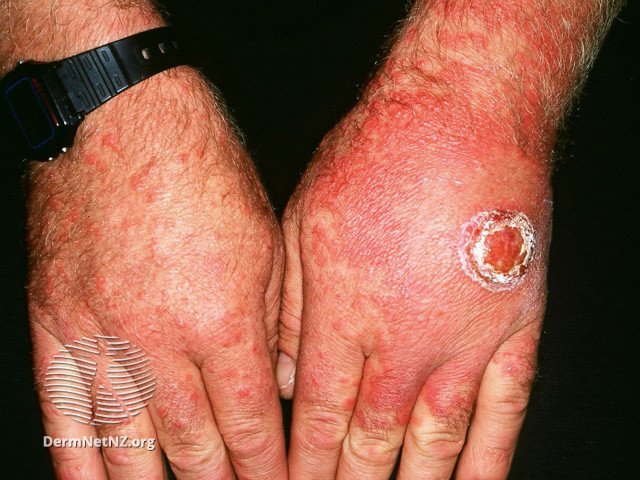
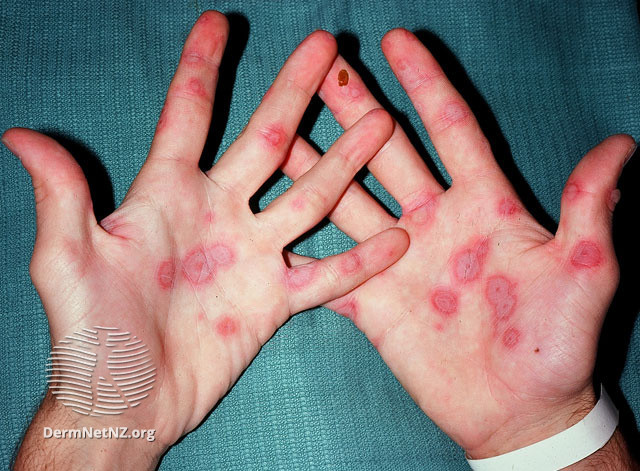
- Cutaneous lesions initially appear on the periphery and then move centrally.
- The distribution is typically symmetrical, with a tendency to favor extensor surfaces.
- May cause pain, itching, or swelling.
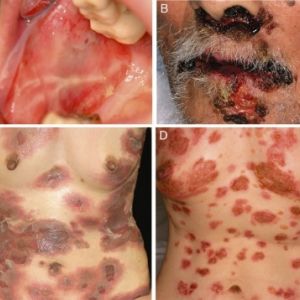
- Initial lesions manifest as circular, red papules that subsequently progress into target-shaped lesions.
Target lesions are characterized by three concentric rings of color variation:
-A dark, central region of epidermal necrosis
-Encircled by a less swollen area
-Surrounded by an erythematous periphery.
Atypical lesions can coexist alongside typical lesions. Atypical lesions exhibit elevated surfaces with indistinct boundaries.
In severe disease, up to hundreds of lesions in varying developmental stages may be present making it challenging to distinguish characteristic lesions.
Mucosal features:
- Lesions form as blisters, which then rupture to reveal superficial erosions covered by a white pseudo-membrane.
- Erythema multiforme primarily affects the oral membranes, but can also manifest with urogenital and, infrequently, ocular lesions.
- When the mucous membrane is affected, it can cause pain and greatly restrict the ability to eat; these lesions may occur before or after skin lesions.
The symptoms are anticipated to resolve spontaneously after 4 weeks from the start of the condition (or up to 6 weeks in cases of severe disease).
Diagnosis:
The diagnosis of acute erythema migrans (EM) is often established by evaluating the patient’s history and thorough physical examination of their clinical symptoms. Skin biopsies are helpful in confirming the diagnosis when there is uncertainty. Direct immunofluorescence studies may be necessary to rule out the presence of autoimmune blistering disease.
Differential diagnosis:
- Urticaria
- Viral exanthem
- Stevens-Johnson syndrome
- Fixed drug eruption
- Bullous pemphigoid
- Paraneoplastic pemphigus
- Polymorphous light eruption
- Rowell syndrome (erythema multiforme-like lesions in systemic lupus erythematosus)
Management:
Treatment is frequently unnecessary as episodes are generally self-limiting without any persistent complications. However, the presence of ocular involvement should always result in a referral to an ophthalmologist due to the potential for more severe consequences.
Management of symptomatic mild cases:
- Itch: To relieve itch or discomfort caused by skin lesions, oral antihistamines and/or topical steroids might be used.
- Pain: For mild mucosal involvement, oral washes containing antiseptic or local anesthetic might be used to relieve pain.
Additional therapies are dependent on the underlying cause:
- Infection: treat precipitating infections appropriately (notice that treating HSV does not change the course of a single episode of erythema multiforme significantly).
- Offending medication: stop any offending medication and avoid in future.
Severe mucosal disease:
- Admission to the hospital might be necessary to support oral intake.
Chronic illness:
- A minimum duration of 6 months of continuous oral antiviral treatment (usually acyclovir/aciclovir), regardless of whether a definite cause has been determined.
- Achieving remission might be challenging and may necessitate trying a longer course of treatment or a different antiviral medication.
Written by:
Mashael Alanazi, Medical Student.
Revised by:
Maee Barakeh, Medical Student.
Resources:
DermNet
UpToDate