Milia

Overview:
Milia are small, superficial, harmless cysts commonly seen across all ages. The term milia refers to multiple lesions, while milium a single lesion. These cysts originate either from the infundibulum of hair follicles or from eccrine ducts. When found in oral cavity, they arise from minor salivary gland ducts.
Clinical Features:
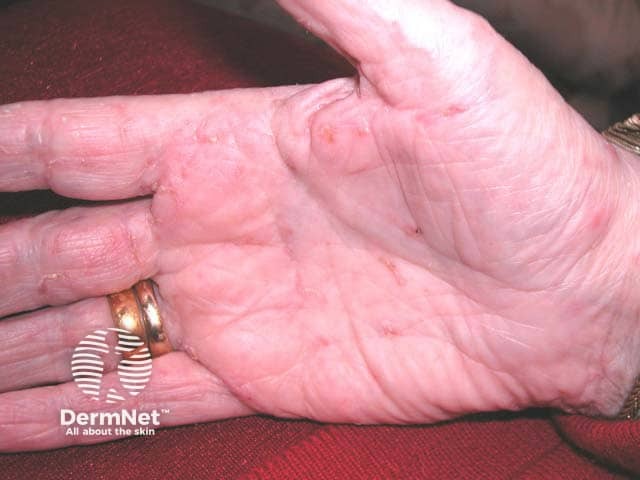
Milia present as firm, white to yellow papules measuring 1-2 mm in diameter. The face, particularly the eyelids, is the most frequently affected area. Several clinical types of milia exist:
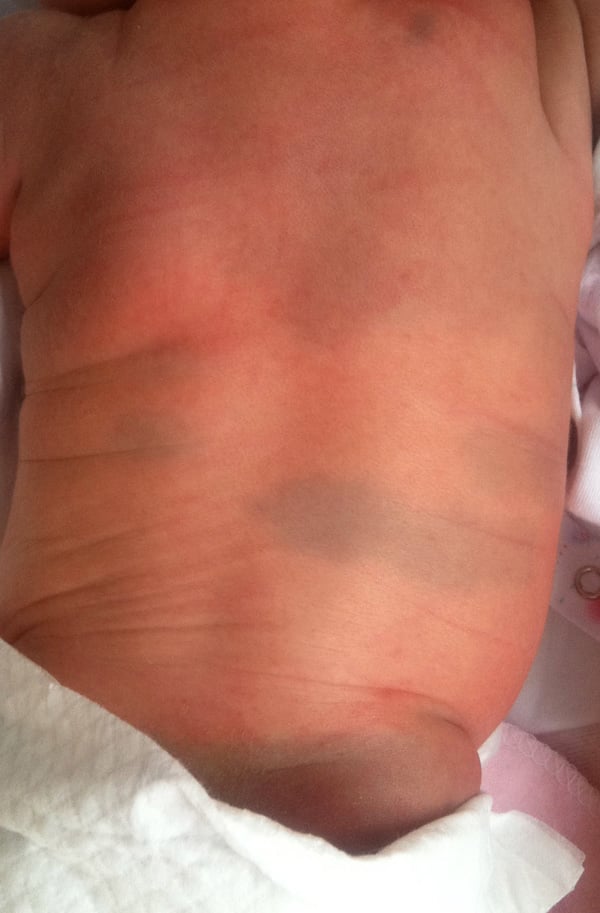
Neonatal milia:
Affect approximately 40-50% of newborn, these lesions may appear in clusters on the nose, scalp, or inside the mouth. They usually resolve spontaneously within the first few weeks of life.
Primary milia in children and adults:
These commonly occur on the eyelids, cheek, forehead, and genitalia.
Juvenile milia:
These may be linked to inherited genetic conditions.
Milia en plaque:
A rare variant appears as a multiple milia within erythematous edematous plaque, most often located postauricular area.
Traumatic milia:
These develop at sites of skin injury, such as thermal burns or blistering rashes. They originate from eccrine sweat ducts.
Drug-induced Milia:
Certain topical medications, including corticosteroids and hydroquinone, have been associated with the development of milia.
Diagnosis:
The diagnosis is primarily clinical and is based on the characteristic appearance of the lesions. In rare or uncertain cases, a skin biopsy may be performed to confirm diagnosis.
Histopathology:
On histological examination, milia are identified as small epidermoid cysts with stratified squamous epithelial lining and a granular layer. The cyst cavity contains laminated keratin.
Treatment:
Milia are benign and often resolve spontaneously within few months. In newborns, milia resolve within 4 weeks of life. For cosmetic reasons or in persistent cases, individual milia may be removed by incising the overlying epidermis using needle, lancet, or scalpel, followed by expressing the cyst contents. Other treatment options include:
- Laser ablation
- Electrodesiccation.
- Topical retinoids – beneficial in cases of multiple milia.
Done by:
Layan Alshehri, Medical Intern
Revised by:
Naif Alshehri, Medical Intern
Resources:
Bolognia 5th Edition
DermNet