Sturge–Weber syndrome
What is Sturge–Weber syndrome?
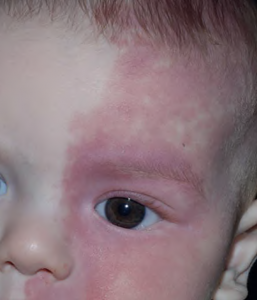
Sturge-Weber syndrome (SWS), also known as encephalotrigeminal angiomatosis, is a neurocutaneous disorder characterized by capillary malformation on the facial skin and capillary-venous malformations in the brain and eyes. A facial cutaneous venous dilatation, often known as a nevus flammeus or port-wine birthmark, is a hallmark of SWS.
Causes of Sturge–Weber syndrome
Sturge-Weber syndrome is caused by a somatic mosaic mutation in the GNAQ gene on chromosome 9. This indicates that the gene mutation occurred in body cells following the formation of the zygote. GNAQ regulates intracellular signaling pathways. In affected cells, the mutation causes uncontrolled formation or maturation of capillaries.
Clinical features of Sturge–Weber syndrome
Sturge-Weber syndrome is marked by vascular malformations on the face as well as in the eye and brain of affected individuals. These are present at birth.
Port-wine stains are the most frequent type of vascular malformation, affecting around three in every 1000 infants, but most are not associated with Sturge–Weber syndrome.
- It is frequently the first component of the disease to be noticed because it is apparent at birth. It may be extremely pale at first, but it normally darkens with age.
- It typically appears on the forehead and upper eyelid, where the first and second divisions of the trigeminal nerve run.
- It can also impact both sides of the face. Leptomeningeal vascular malformations arise inside the brain on the same side as the port-wine stain. It may also occur without a port-wine stain.
Neurological and ophthalmological symptoms are progressive and typically appear in the first two years of life. These may include:
- Seizures and Epilepsy
- Hemiparesis and stroke-like events
- Behavioral problems
- Visual field defects and glaucoma.
- Growth hormone deficiency.
Diagnosis of Sturge–Weber syndrome
Sturge-Weber syndrome is diagnosed by identifying the characteristic trigeminal port-wine stains and leptomeningeal capillary-venous malformations. A diagnosis based on leptomeningeal lesions alone depends on the development of symptoms.
If neurological symptoms or abnormalities are present, magnetic resonance imaging (MRI) of the brain with gadolinium contrast is performed to detect leptomeningeal capillary-venous malformations.
Differential diagnosis of Sturge–Weber syndrome:
Here is a list of vascular malformation syndromes with its characteristics:
- Klippel–Trénaunay syndrome is characterized by extensive capillary malformations that primarily affect the limbs and trunk, often leading to hypertrophy of the involved limb.
- Proteus syndrome manifests as asymmetrical and disproportionate overgrowth of various body parts, caused by a somatic activating mutation in the AKT1 oncogene.
- Parkes–Weber syndrome features large capillary malformations on extremities, accompanied by hypertrophy and multiple fast-flowing arteriovenous shunts.
- CLOVES syndrome is identified by congenital lipomatous overgrowth, vascular malformations, epidermal nevi, and spinal or skeletal anomalies, including scoliosis. It results from somatic mosaic activating mutations in the PIK3CA gene.
Treatment of Sturge–Weber syndrome
Sturge-Weber syndrome has no specific treatment. Treatment focuses on managing the cutaneous, neurological, and ocular symptoms.
- Treatment of seizures in patients with Sturge–Weber syndrome often proves challenging, as antiepileptic drugs do not always yield effective results. Currently, no specific treatment has been identified as consistently superior to others.
- Port-wine stains associated with the condition can be addressed using pulsed-dye laser therapy, although the outcomes are variable. Given the generally limited success of pulsed-dye lasers alone, adjunctive treatments involving topical antiangiogenic agents are being trialled.
- Regular ophthalmologic evaluations are advised due to the frequent occurrence of glaucoma in Sturge-Weber syndrome, which is managed through both surgical and medical interventions.
- Infants diagnosed with Sturge–Weber syndrome should receive low-dose aspirin therapy as this approach may help prevent disease progression by enhancing cerebral blood flow and potentially reducing the risk of neuronal damage.
Written by:
Atheer Alhuthaili , Medical Student.
Revised by:
Naif Alshehri, Medical Intern.
References:
DermNet.
Picture Reference:
Bolognia Dermatology 5th Edition.