Cherry Angiomas

Definition
Cherry angiomas are benign vascular skin lesions characterized by capillary proliferation within the dermis. Clinically known as Campbell de Morgan spots, these papular lesions are among the most common acquired cutaneous vascular proliferations. Though typically asymptomatic, their appearance can be cosmetically concerning. In rare instances, eruptive forms may be associated with systemic conditions.
Epidemiology
Cherry angiomas are extremely common, especially with increasing age. They usually start to appear in adulthood most often after the age of 30 and gradually become more numerous over time.
- By the age of 60, most people will have at least one.
- By 75 years old, around 75% of individuals are affected.
They occur equally in men and women, and affect all ethnic groups, although they tend to be more visible in lighter skin tones. There may also be a genetic tendency, as some people have a family history of similar lesions.
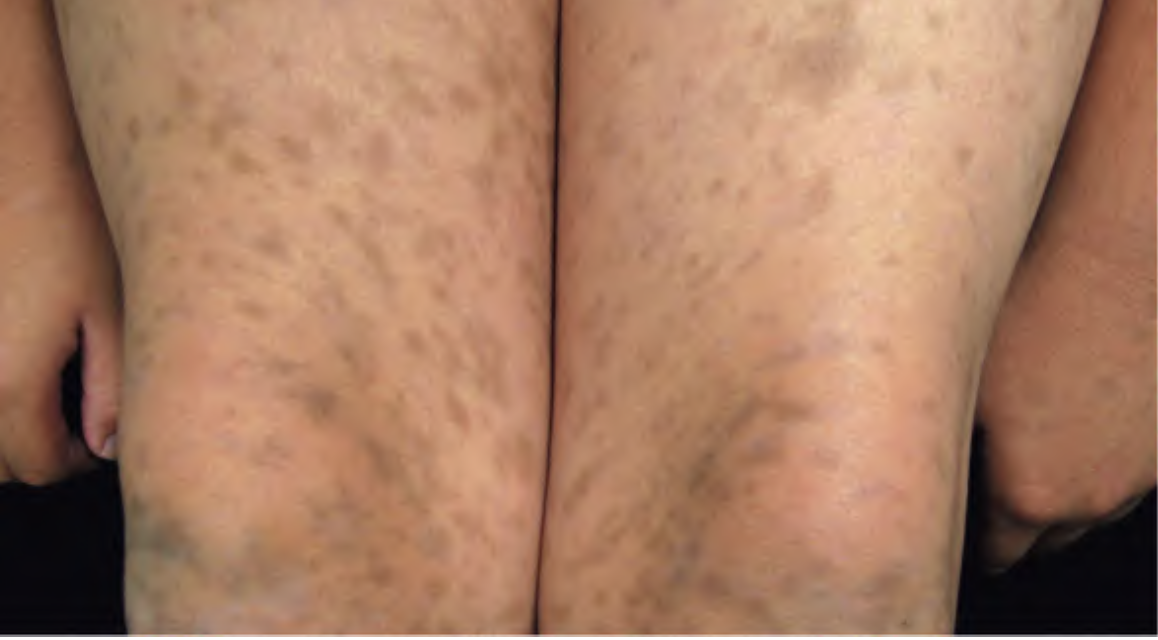
Rarely, sudden onset of numerous lesions (eruptive cherry angiomas) has been reported in association with internal malignancy or during pregnancy.
Etiology and Pathogenesis
Exact cause is not fully known, but several factors are linked:
- Hormonal factors
– More common during pregnancy
– May shrink after delivery
– Linked to high prolactin levels in some cases
- Genetic mutations
Changes in certain genes like GNAQ, GNA11, and GNA14, Also sometimes seen in HRAS and KRAS genes. These genes are involved in blood vessel growth.
- Possibly minor trauma or skin irritation:
May trigger development in some cases.
- Not caused by cancer, but may rarely appear alongside internal tumors or systemic diseases
Clinical Features
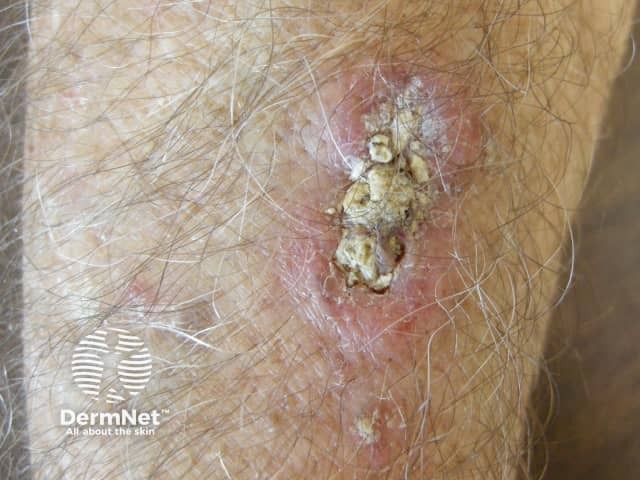
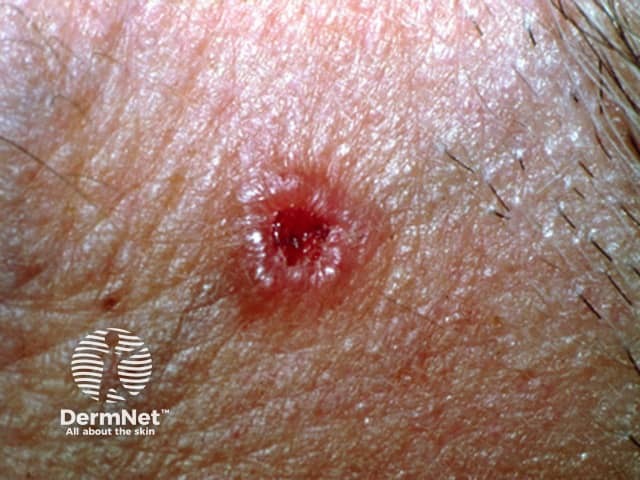
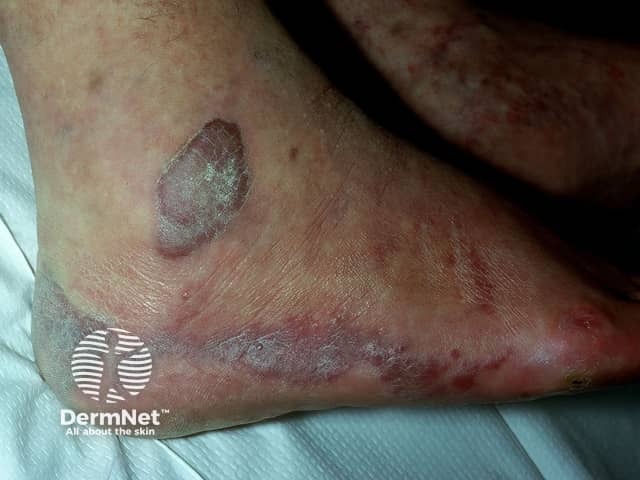
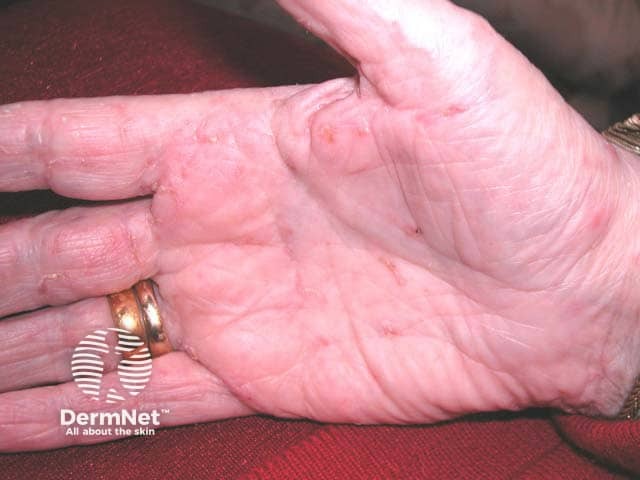
Cherry angiomas present as firm, dome-shaped papules ranging from red to blue or purple in color, typically measuring 1–10 mm in diameter. They are usually multiple and randomly distributed across the trunk and extremities, sparing mucous membranes, hands, and feet. The lesions are asymptomatic but may bleed if traumatized. In thrombosed angiomas, the surface may appear blackish, though dermoscopy can help reveal the underlying vascular hue.
Diagnosis
Diagnosis is primarily clinical, supported by characteristic dermoscopic features, such as red to purple lacunae in a lobular or “red clod” pattern. Histological evaluation, when necessary, reveals dilated venules within a thickened papillary dermis with intervening collagen bundles. Cherry angiomas should be distinguished from petechiae and glomeruloid hemangiomas, particularly when multiple lesions erupt suddenly or there is suspicion of systemic disease.
Management
Cherry angiomas are benign and generally require no treatment unless they are symptomatic, subject to recurrent trauma, or cosmetically undesirable. When removal is indicated, options include:
– Laser therapy (e.g., pulsed dye laser)
– Electrosurgery
– Cryotherapy
– Shave excision
Recurrence following treatment is uncommon, and procedures are typically well-tolerated.
Written by:
Raneem Alahmadi, Medical Intern.
Revised by:
Naif Alshehri, Medical Intern
Resources:
Bolognia 5th edition
Dermnet