Neurofibromatosis type 1
Definition and epidemiology:
Neurofibromatosis type 1, also called Von Recklinghausen’s disease, is a genetic disorder characterised by cutaneous as well as peripheral and/or central nervous system neoplasms. Neurofibromatosis type 1 incidence rate is approximately 1 in 3000 births.
Pathogenesis:
Neurofibromatosis type 1 is inherited in an autosomal dominant pattern. 50% of patients have de novo mutations that occur in paternally derived chromosomes.
NF1 is caused by pathogenic variants of NF1 gene, located in chromosome 17q11.2. The gene encompasses a protein called neurofibromin.
Clinical features:
The clinical features of NF1 are myriad and vary in frequency.
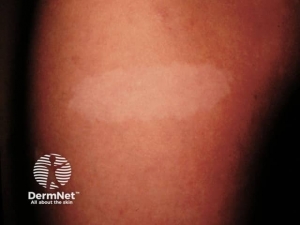
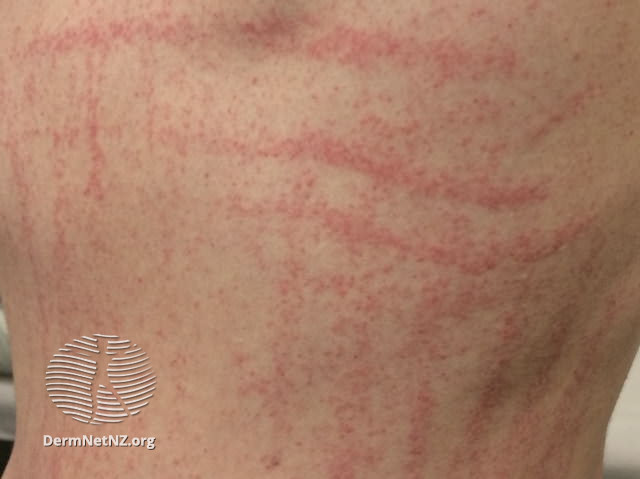
Café-au-lait macules– are flat, uniformly hyperpigmented macules that appear during the first year after birth and usually increase in number during early childhood . The number of café-au-lait macules then stabilizes over time. Up to 15 percent of the general population has one to three café-au-lait macules; however, the presence of six or more café-au-lait macules is highly suggestive of NF1. Approximately 95 percent of adults with NF1 have café-au-lait macules, but they tend to fade later in life and may be difficult to distinguish in older individuals.
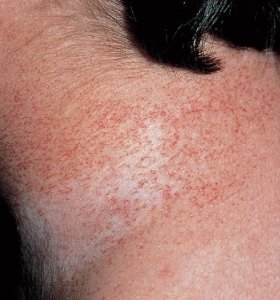
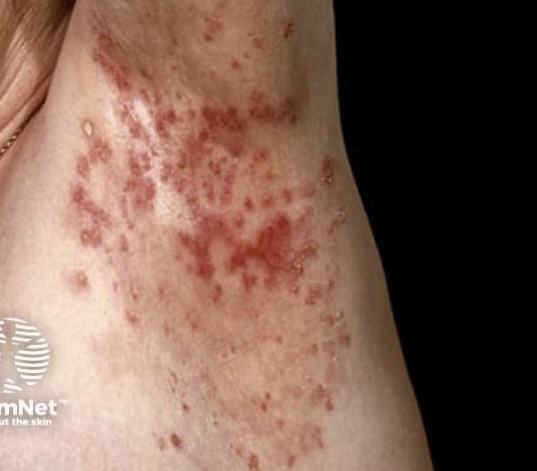
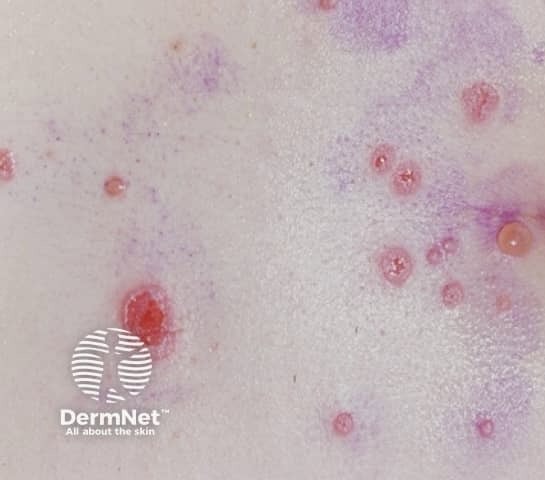
Freckling– Freckling in the axillary or inguinal regions (Crowe sign) is a diagnostic criterion distinct from café-au-lait macules. Freckling usually is not apparent at birth but often appears by age three to five years, typically first in the inguinal region.
Lisch nodules – Lisch nodules are raised, tan-coloured hamartomas of the iris and represent a specific finding for NF1.
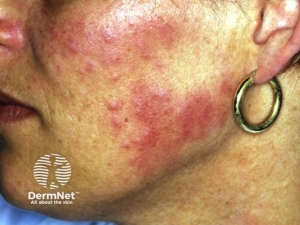
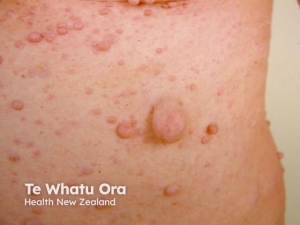
Neurofibromas – Neurofibromas are the most common type of benign tumour that develops in people with NF1. Neurofibromas are benign peripheral nerve sheath tumours that are composed of a mixture of Schwann cells, fibroblasts, perineurial cells, mast cells, macrophages, and T cells.
- Cutaneous neurofibromas: are skin-coloured to pink, tan, or brown papulonodules that are soft or slightly rubbery in texture and can range from a few millimetres to several centimetres in diameter. While fully formed CNFs are often dome-shaped or pedunculated, early lesions may be barely elevated. They invaginate easily with gentle pressure, exhibiting the pathogno- monic “buttonhole” sign.
- Subcutaneous neurofibromas occur deeper in the dermis and subcutis, tending to be firmer and less well-circumscribed than cutaneous neurofibromas.
- Plexiform neurofibromas may track along nerves to create tender, firm nodules or masses in the subcutaneous tissue with a “bag of worms” consistency upon palpation.
Neurological manifestations – these include optic gliomas, learning difficulties, speech problems and behavioural issues.
Skeletal manifestations – Macrocephaly, hypertelorism, scoliosis, and osteopenia are common cranioskeletal abnormalities in individuals with NF1. A highly distinctive but less common finding is pseudarthrosis of the long bones, most often the tibia. Sphenoid wing dysplasia is a congenital, typically unilateral bony defect in the posterior wall of the orbit.
Cardiovascular manifestations – Hypertension is a common finding in NF1 patients. Although essential hypertension is most frequent, renovascular stenosis (especially in children) and unsuspected pheochromocytomas may be the cause in some patients. Pulmonary stenosis can occur in 1% of NF1 patients.
Diagnosis:
The diagnosis of NF1 is based upon the presence of characteristic clinical features. Genetic testing is not required to make the diagnosis but can be helpful in establishing the diagnosis in children who do not meet diagnostic criteria.
Diagnostic criteria – Two or more of the following must be present:
- Six or more café-au-lait macules >5 mm in prepubertal individuals and >15 mm in postpubertal individuals.
- Two or more neurofibromas of any type or one plexiform neurofibroma.
- “Freckling” in the axillary or inguinal regions.
- Optic gliomas.
- Two or more Lisch nodules (iris hamartomas).
- Osseous lesions, such as sphenoid wing dysplasia or thinning of long bone cortex, with or without pseudarthrosis .
- First-degree relative (parent, sibling or offspring) with NF1 by the above criteria.
Management:
Management of patients with NF1 requires a multi-disciplinary approach. Dermatologists are often consulted to assist in establishing the diagnosis in affected children. Goals of longitudinal care include early recognition and treatment of complications, maximization of academic and vocational achievement, and minimization of the disease’s psychosocial impact.
Plexiform neurofibromas (PNF) may grow into large, bulky tumors during early childhood. Multidisciplinary teams are often required to excise deep and extensive neurofibromatous tissue, and the lesions tend to recur following surgery.
Café-au-lait macules do not need or respond well to treatment, although it may be desired for cosmetic purposes. When laser therapy is performed, CALMs tend to persist or recur.
Lifelong surveillance for the development of Malignant peripheral nerve sheath tumors (MPNST) is essential, particularly for patients at increased risk of this malignancy due to the known presence of PNFs, polyneuropathy secondary to multiple nerve root tumors, a history of radiation therapy, or a germline NF1 microdeletion. PET- CT scan can aid in the early detection of malignant change within Plexiform neurofibromas.
Written by:
Bandar Alharbi, Medical Intern
Revised by:
Naif Alshehri, Medical Intern
Resources:
Dermnet
UpToDate
Bolognia Dermatology textbook