Harlequin Ichthyosis

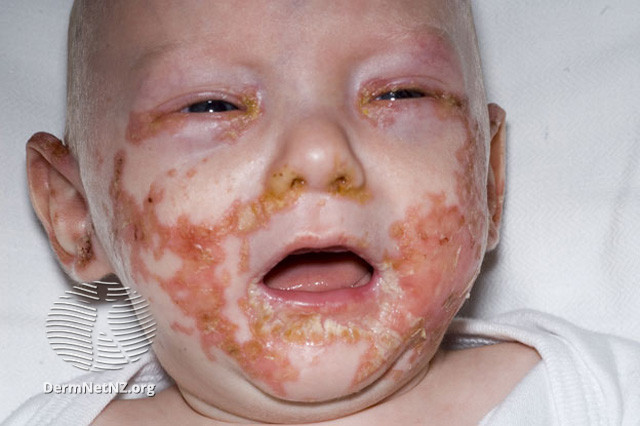
A severe genetic variant of ichthyosis called harlequin ichthyosis manifests at birth as thicker, rigid skin plates that cover the entire body like armor. The condition is uncommon and equally affects men and women. Approximately one in 300,000 neonates are affected by it. Harlequin ichthyosis is not known to be racially predisposed. There could be a higher occurrence in societies where paternal consanguinity is prevalent.
Causes:
Harlequin ichthyosis is inherited in an autosomal recessive manner. The ABCA 12 gene alterations are to blame. It is thought that the ABCA12 gene encodes a transporter protein that is necessary for healthy skin development and is involved in the movement of epidermal lipids across cell membranes. The cell cannot produce any ABCA12 protein due to certain mutations in the ABCA12 gene. Additional mutations result in the synthesis of an atypically tiny protein that is incapable of carrying lipids in an appropriate manner. Harlequin ichthyosis is characterized by hard, thick scales that are caused by a disruption in the normal development of the epidermis caused by a loss of functional ABCA12 protein.
Clinical features:
The clinical features include skin changes present at birth, significantly thickened, with deep erythematous fissures separating the scales and huge, glossy plates of hyperkeratotic scale. Facial features include degraded nasal alae and blocked nares. The external auditory canal may be blocked by scale, and the ears’ pinnae and retroauricular folds may be tiny or nonexistent. The lips’ traction results in eclabium.
Diagnosis:
Genetic laboratory testing and a physical examination of the patient are necessary for the diagnosis of harlequin ichthyosis. The most precise diagnostic procedure for harlequin ichthyosis involves looking for a loss of function mutation in the ABCA12 gene using genetic testing. Gene mutations may result in smaller versions of the proteins essential for skin development as well as poor lipid transport in the skin. A collodion membrane and congenital ichthyosiform erythroderma-like appearance are the outcomes of less severe mutations. Histology of the skin biopsy reveals hypergranulosis, parakeratosis, and a significantly thicker stratum corneum.
Management:
Treatment for harlequin ichthyosis focuses on skin protection and infection control; there is no known cure. For several weeks during their early years, the majority of harlequin babies require one-on-one nursing care. Infection prevention or treatment during this period may also require antibiotics. After bathing, when the skin is still damp, softening emollients especially those that contain urea, salicylic acid, or alpha hydroxy acids work exceptionally well. These products help to maintain the skin’s suppleness and moisture content while guarding against the fissures and cracks that can result in a subsequent bacterial infection. It has also been demonstrated that early systemic treatment with oral retinoids (such as acitretin or isotretinoin) improves overall survival, softens, or resolves plate-like scales, and heals skin fissures.
Complications:
Poor immunity, infections, muscle shortening, limb necrosis and amputation, and respiratory failure.
Outcome:
Infants with harlequin ichthyosis in the past hardly made it past the first few days of life. The survival rate has increased due to innovations in newborn care; globally, rates are getting close to 50%. Youngsters who make it through the neonatal stage typically acquire a less severe form of ichthyosis, but they still exhibit fish-scale scales, seborrheic patches that retain waxy, yellowish material, generalized poor hair growth, and scarring alopecia. Those who survive harlequin ichthyosis may nevertheless experience hypothyroidism, small stature, arthralgias, digit contractures, and failure to flourish.
Written by:
Mohammed Alahmadi, Medical Student.
Revised by:
Maee Barakeh, Medical Student.
References:
DermNet
Dermatology by Bolognia
Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology
Image source: Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology