Anaphylaxis
Definition:
Anaphylaxis is an acute, potentially lethal, multi system syndrome resulting from the sudden release of mast cell- and basophil-derived mediators into the circulation. It most often results from immunologic reactions to foods, medications, and insect stings, although it can also be induced through non immunologic mechanisms by any agent capable of producing a sudden, systemic degranulation of mast cells or basophils.
Pathophysiology:
The World Allergy Organization (WAO), an international umbrella organization representing a large number of regional and national professional societies dedicated to allergy and clinical immunology, has proposed discarding this nomenclature. The WAO categorizes anaphylaxis as either immunologic or nonimmunologic.
1- Immunological Anaphylaxis:
The classical mechanism associated with human allergic disease is initiated by an antigen (allergen) interacting with allergen-specific IgE bound to the receptor on mast cells and/or basophils.
B cells are driven to differentiate into IgE-producing cells via the activity of the type 2 subset of CD4-bearing helper T cells (Th2 cells). This process largely takes place in the peripheral lymphoid tissues. The cytokines interleukin (IL) 4 and and IL-13 contribute to IgE responses in humans.
Once produced, allergen-specific IgE diffuses through the tissues and vasculature and constitutively occupies high-affinity IgE receptors (Fc-epsilon-RI) on mast cells and basophils.
When allergen diffuses into the proximity of a mast cell or basophil, it interacts with any surface-bound IgE that is specific for that allergen. If signaling is sufficiently robust, the mast cell (or basophil) becomes activated and degranulates, releasing preformed mediators, enzymes, and cytokines (such as histamine, tryptase, and tumor necrosis factor [TNF], respectively) and initiating additional mediator, cytokine, and enzyme production.
2- Non Immunological Anaphylaxis:
Anaphylactic reactions to various drugs have revealed potential mechanisms by which mast cells and basophils could be activated without evidence of involvement of IgE, other antibodies, or immune complexes.
Clinical features:
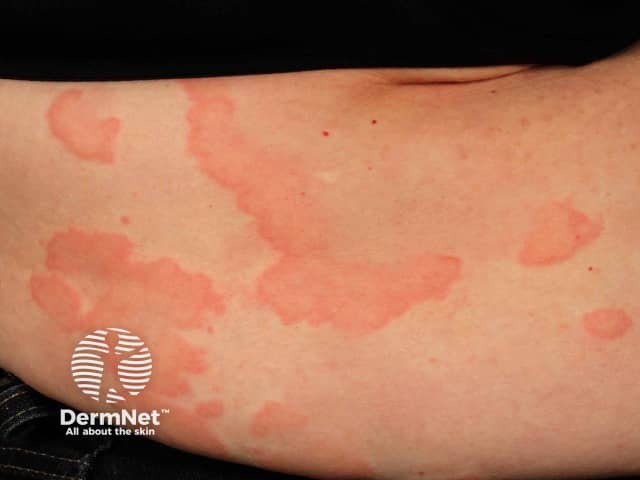
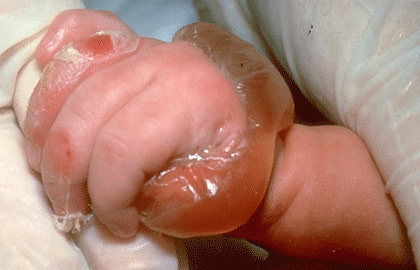
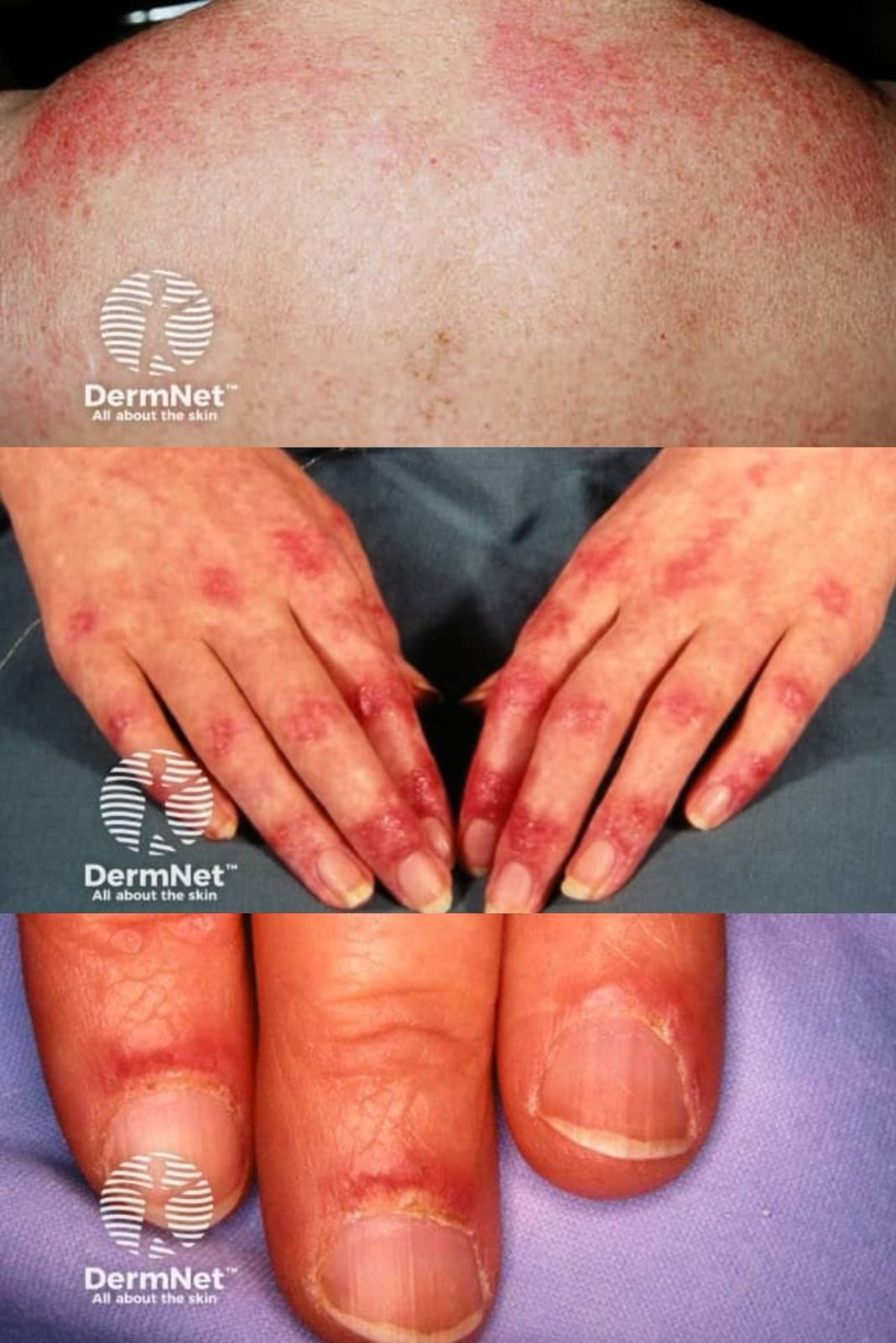
Skin and mucosal involvement:
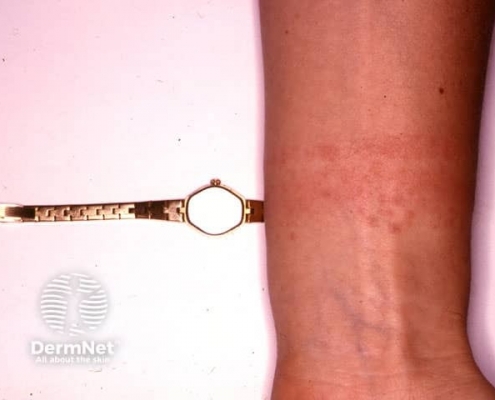
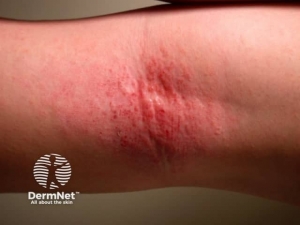
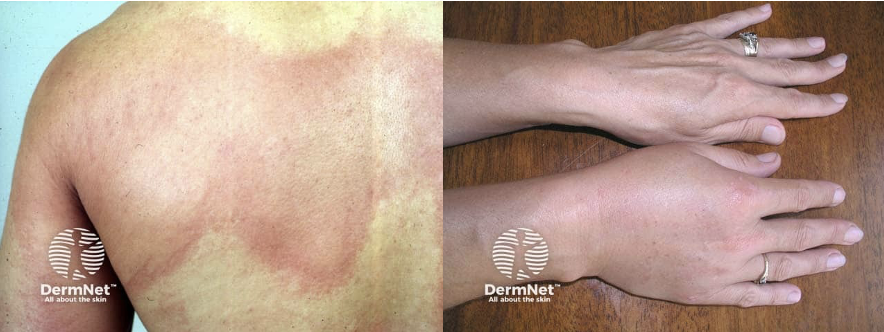
- Generalized hives
- Itching
- Flushing
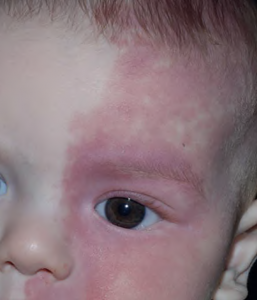
- Swollen lips, tongue or uvula
- Periorbital edema
Respiratory:
- Nasal congestion
- Sneezing
- Throat itching
- Sensation of throat closure or choking
- Wheezing
- Shortness of breath
Gastrointestinal:
- Nausea and vomiting
- Crampy abdominal pain
- Diarrhea
Cardiovascular:
- Syncope
- Dizziness
- Hypotension
- Tachycardia
Diagnosis:
Because acute anaphylaxis can be immediately life-threatening, the diagnosis must be made quickly and efficiently, often while administering initial medication. Diagnosis is essentially made based on:
- Sudden onset of typical symptoms and signs, involving at least two organ systems (hypotension, airway swelling, wheeze, urticaria)
- Development of specific symptoms after exposure to a known or likely allergen
- Exclusion of other diseases that may have similar signs and symptoms.
Management:
Acute anaphylaxis must be treated as a medical emergency with stabilisation of airway, breathing and circulation. Intramuscular epinephrine 0.3–0.5 mg (0.3–0.5 mL of 1:1000 using an autoinjector or syringe) must be given immediately to adult patients with signs of shock, airway swelling, or definite difficulty in breathing. The epinephrine is repeated after 5 minutes if there has been no improvement in hypotension, airway swelling, or wheeze.
Agents that may be given as adjunctive therapies to epinephrine in the treatment of anaphylaxis include H1 antihistamines, H2 antihistamines, bronchodilators, and glucocorticoids. None of these medications should be used as initial treatment or as sole treatment, because they do not relieve upper or lower respiratory tract obstruction, hypotension, or shock and are not lifesaving.
To reduce the risk of recurrence, patients who have been successfully treated for anaphylaxis subsequently require confirmation of the anaphylaxis cause as well as anaphylaxis education. All those at risk of anaphylaxis should wear a Medic alert/emergency bracelet with full details of allergies and contact details of their doctor. In some cases, a patient or caregiver should always carry an emergency kit containing self-injectable Epinephrine and antihistamine tablets.
Written by:
Bandar Alharbi, Medical Intern
Revised by:
Naif Alshehri, Medical Intern
Resources:
UpToDate
Dermnet
Ministry of health
Mayo clinic