What is Tinea Versicolor?
Tinea versicolor is a commonly occurring, benign, superficial cutaneous fungal infection that is typically characterized by patches and macules on the back and chest that are either hyper- or hypopigmented. It is caused by Malassezia species. Despite the availability of efficient oral and topical treatment methods, disease recurrence is frequent.
Epidemiology of Tinea Versicolor
Tinea versicolor occurs worldwide, with a higher occurrence in tropical climates. Adolescents and young adults are the most common age groups to develop tinea versicolor. It can also impact children and infants.
Risk Factors for Tinea Versicolor
There are various risk factors associated with this disease. These risk factors include exposure to hot and humid weather, hyperhidrosis, use of topical skin oils, genetic predisposition, immunosuppression state, malnutrition, corticosteroid usage, and Cushing disease. It may clear in the winter and reoccur each summer.
Cause of Tinea Versicolor
Tinea versicolor does not result from a dermatophyte infection. It is considered non-contagious. The causative organism is a Malassezia species. It is a component of the normal skin flora, but it can cause the disease when it transforms into its pathogenic hyphal form. The reasons for the transformation of tinea versicolor are likely to be multifactorial.
Clinical Features of Tinea Versicolor
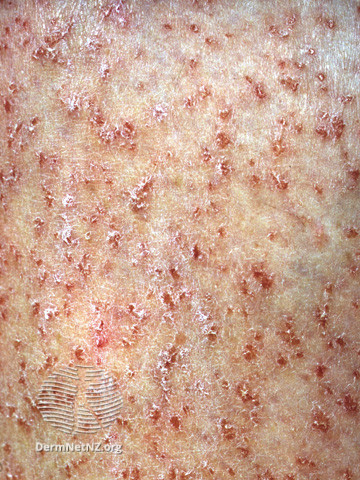
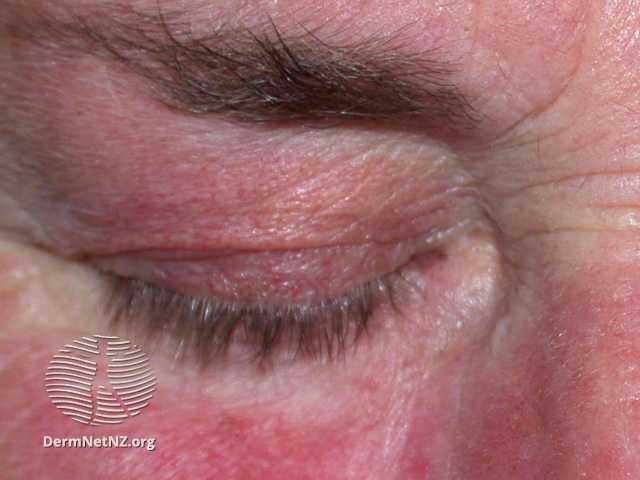
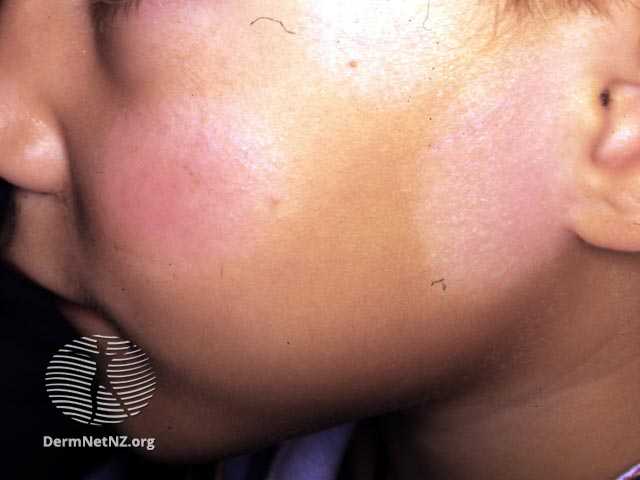
Tinea versicolor is typically asymptomatic; some complain of mild pruritus, with the main issue being its appearance.Tinea versicolor lesions are characterized by hyperpigmented, hypopigmented, or erythematous patches, macules, or thin plaques with a mild scale. To demonstrate this scale, the skin surface may need to be scratched. These lesions are usually small (<15 mm) and range in color from coppery brown, paler than the surrounding skin, or pink. The most typical areas of involvement are the upper trunk and proximal upper extremities. Less frequently, faces (common in children) or intertriginous areas are affected.
Diagnosis of Tinea Versicolor
Tinea versicolor is usually diagnosed clinically. Dermatoscopy and Wood’s lamp examination may show signs suggestive of tinea versicolor but do not confirm the diagnosis. Dermoscopic examination reveals fine-scale, nonuniform pigmentation within lesions and perilesional hyperpigmentation or hypopigmentation. Wood’s lamp examination exhibits yellow to yellow-green fluorescence in certain patients. The KOH preparation findings in tinea versicolor are considered diagnostic. It shows hyphae and yeast cells that resemble spaghetti and meatballs.
Management of Tinea Versicolor
The management of Tinea versicolor can be summarized into a few fundamental aspects:
1:Patient education
2: Topical antifungals, which are considered first-line therapy, including Selenium sulfide, zinc pyrithione, azoles (miconazole or ketoconazole), or allylamines (terbinafine)
3: Oral antifungals are only for infections that are severe, extensive, or resistant to topical treatment (not commonly used in children). Oral itraconazole and fluconazole are the recommended oral medications.
Recurrence of Tinea Versicolor
Tinea versicolor has a high recurrence rate. Preventive treatment with topical or oral medication administered intermittently is required to avoid recurrences in most cases.
Written by:
Atheer Alhuthaili , medical student.
Revised by:
Maee Barakeh, medical student.
References:
Bolognia textbook of dermatology.
DermNet.
UpToDate.
Medscape .