Sunscreen
Ultraviolet radiation exposure causes deleterious effects on the skin, contributing to skin aging, cancer and photosensitivity. To prevent its impact, sunscreen application is imperative in protecting the skin against these harmful radiation.
The sun emits three main UV radiation at different wavelengths:
- Ultraviolet A (UVA) comprises 95% of the UV radiation. Its intensity is consistent throughout the day and from season to another.
- Ultraviolet B (UVB) makes up the remaining 5% UV radiation detected on the earth’s surface. The intensity of UVB is highest during summer and from 10 a.m to 4 p.m during the day.
- Ultraviolet C (UVC) is absorbed by earth’s atmosphere and is of little concern.
Sunscreen works by absorbing or scattering UV radiation, primarily UVA and UVB, minimizing the effect on the skin. There are two categories of sunscreen agents with different mechanisms: Chemical (organic) and physical (inorganic).
Chemical (organic) filters
It acts by absorbing high energy light. They are further classified into UVA filters and UVB filters but companies use a combination of both filters to yield higher sun protection factor (SPF), stability and broad-spectrum absorption. In addition, organic filters have outstanding safety and aesthetic properties with minimal phototoxicity, photosensitivity and staining on skin.
Physical (inorganic) filters
Inorganic filters reflect and scatter light. Most common agents are metal oxides titanium dioxide and zinc oxide, it’s chemically inert and protects against the full UV spectrum. However the inorganic leaves a whitecast that raises cosmetic concerns.
There are other measures for protection that can be implemented alongside sunscreen application, like choosing long sleeves and trousers, wearing broad hats and adequately applying/reapplying sunscreen.
The use of sunscreen is beneficial in mitigating the risks of skin cancer and disorders from UV radiation. Both chemical and physical sunscreens generally have an excellent safety profile, but chemical sunscreens are systemically absorbed. There are no harms, but further studies are required to confirm that.
Written by: Naif Alalshaikh, medical student.
References:
“Topical Sunscreen Agents.” Sunscreens, Sunblocks | DermNet NZ, https://dermnetnz.org/topics/topical-sunscreen-agents.
Gasparro, Francis P., et al. “A Review of Sunscreen Safety and Efficacy.” Photochemistry and Photobiology, vol. 68, no. 3, 1998, pp. 243–256., https://doi.org/10.1111/j.1751-1097.1998.tb09677.x.
Ngoc, et al. “Recent Trends of Sunscreen Cosmetic: An Update Review.” Cosmetics, vol. 6, no. 4, 2019, p. 64., https://doi.org/10.3390/cosmetics6040064.

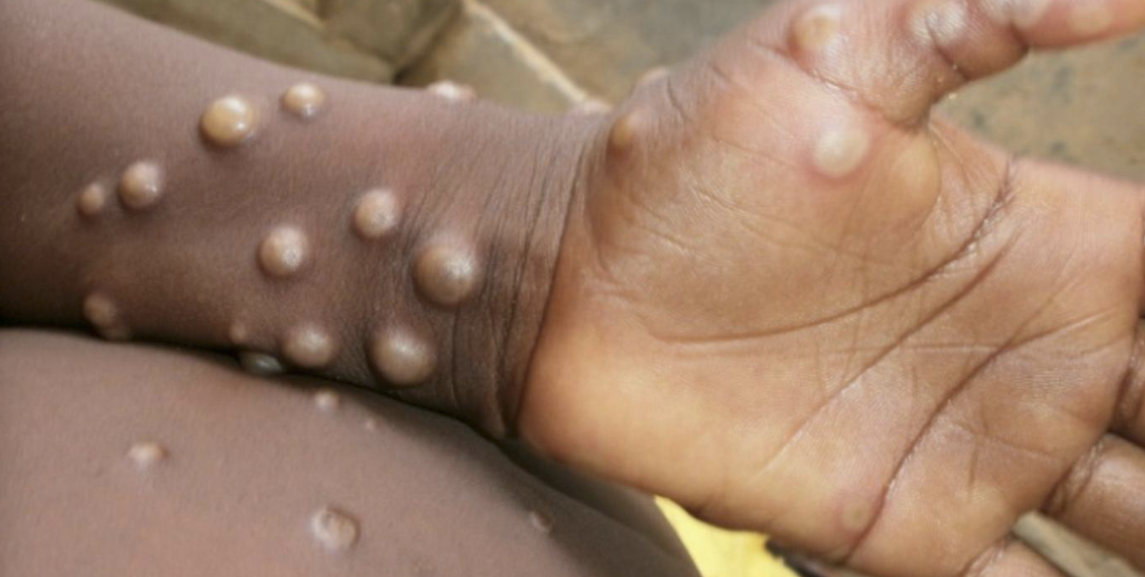

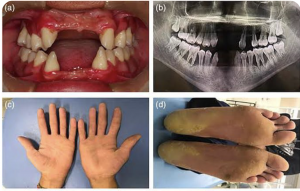
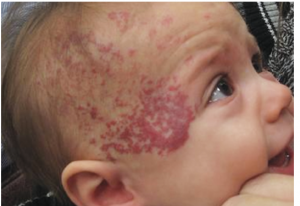
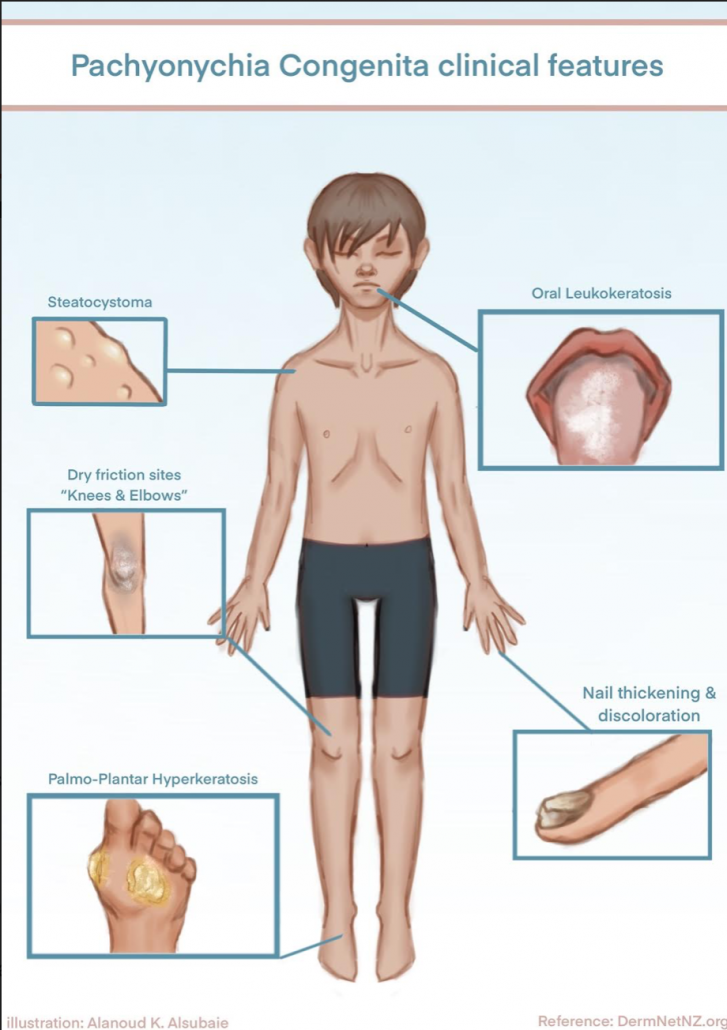 Pachyonychia congenita is a rare inherited disorder of keratinization that primarily affects the nails and skin. Affected people develop thickened skin on the soles and palms, white patches on the tongue and mouth, and bumps around the elbow and knees.
Pachyonychia congenita is a rare inherited disorder of keratinization that primarily affects the nails and skin. Affected people develop thickened skin on the soles and palms, white patches on the tongue and mouth, and bumps around the elbow and knees.